Abstract
There have been several reports of extreme clotting post mRNA vaccination. Many images of these clots have circulated on social media and have drawn scrutiny and accusations of these clots being manufactured in a lab or taken from animals to confuse the public on this important topic. In order to confirm these clots were in fact human derived we performed qPCR of the human house keeping gene RNaseP and followed that up with whole genome sequencing. Both qPCR and Illumina whole genome sequencing confirm this is in fact human DNA. Human DNA is commonly found in clots due to a process known as Neutrophil Extracellular Traps or NETs. Evidence of these NETs are reflected in the sequencing library. 27 clotting and thrombosis related genes were examined for variants that may predispose the patient to this phenomenon. Several moderate compounding mutations are found in Factor V, Factor XIIIB, Factor 7, and GP1BA. An additional 2312 genes with Gene Ontology annotations related to thrombosis were examined for variants of high impact with snpEFF. Several variants in SERPINs, CPB2, and MASP1 were identified in clots from 2 different individuals. Finally, 201 genes with Gene Ontology annotations related to amyloidogenesis were examined for high impact variants. Variants in PSEN2 and LRP1 were identified.
Introduction
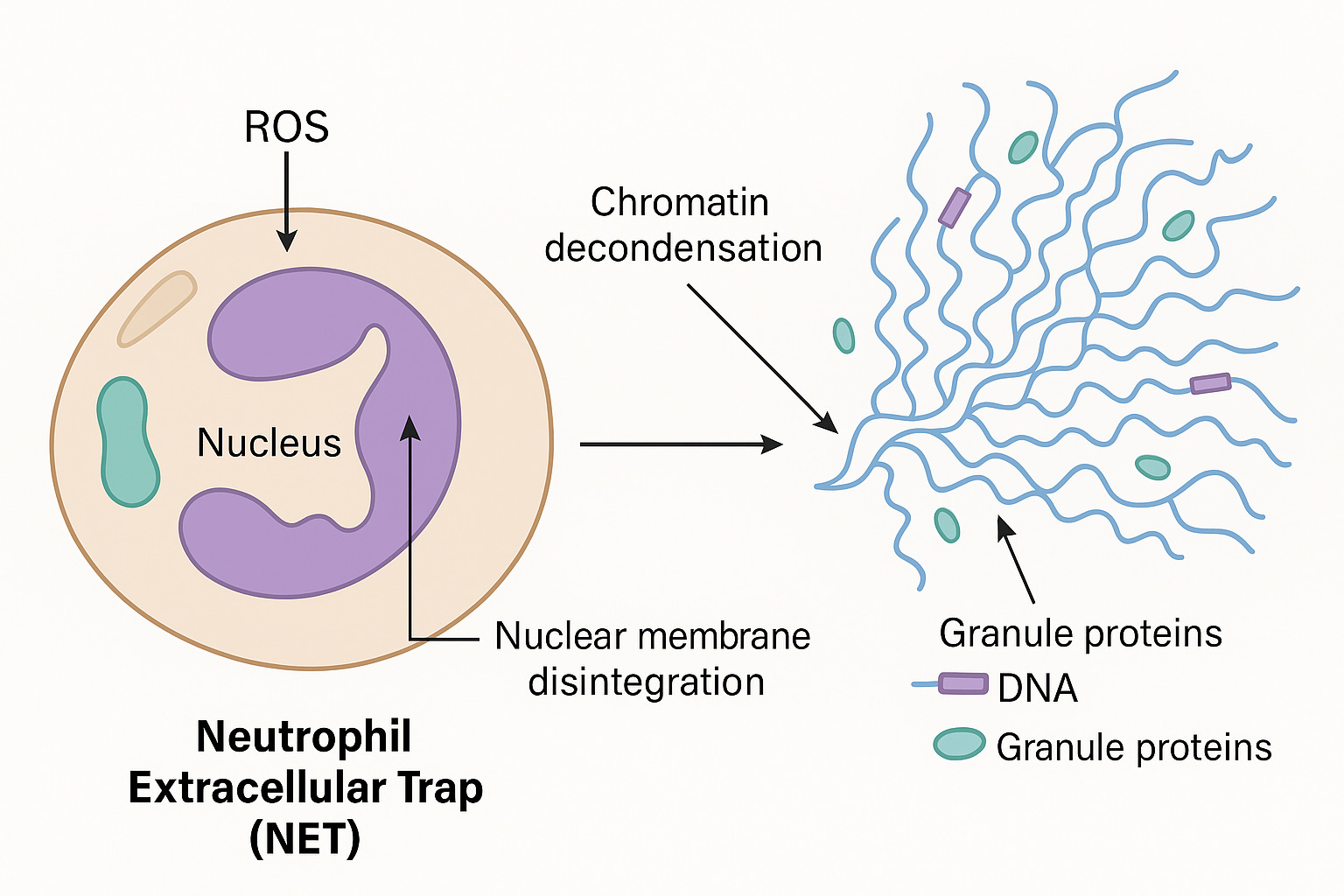
Clotting and thrombosis are often catalyzed by circulating free DNA (cfDNA), Lipopolysaccharides (LPS) and amyloidogenic peptides. All 3 of these components may be present in mRNA vaccines given the E.coli based manufacturing of the plasmids in E.coli and the known amyloidogenic nature of the spike protein. cfDNA is often used as a scaffold in Neutrophil Extracelluar Traps or NETs.


The foundational Brinkman et al paper on NETs is linked here.
A few things to note. This NETs DNA is usually chromatin wrapped with nucleosomes and often times bacteria or other pathogens are entrapped in NETs.
Back in 2008, I was involved in a study with the Nobel laureate Andrew Fire to explore nucleosome positioning on DNA. This study took the cover of Genome Research and was the first peer reviewed paper to utilize the SOLiD sequencing method we developed.

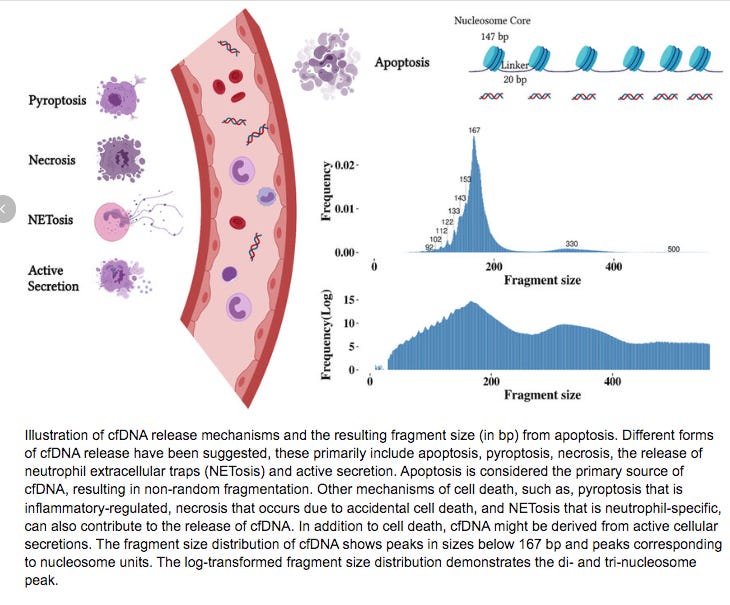
Nucleosomes are protein spools that wrap DNA. They have some physical constraints related to structure of DNA. In B-DNA, its helix has 10.5 bases per turn. This in turn has an impact on how tightly you can wrap DNA around a protein.

DNA that is wrapped around nucleosomes has certain length constraints and often leaves a signature in whole genome sequencing libraries. DNA wrapped in nucleosomes is usually 147 bases in length.

Below you can see the DNA fragment lengths from two clots. This fragment length is precise to the individual base as it is the length of the DNA after it has been sequenced and mapped to the human genome.
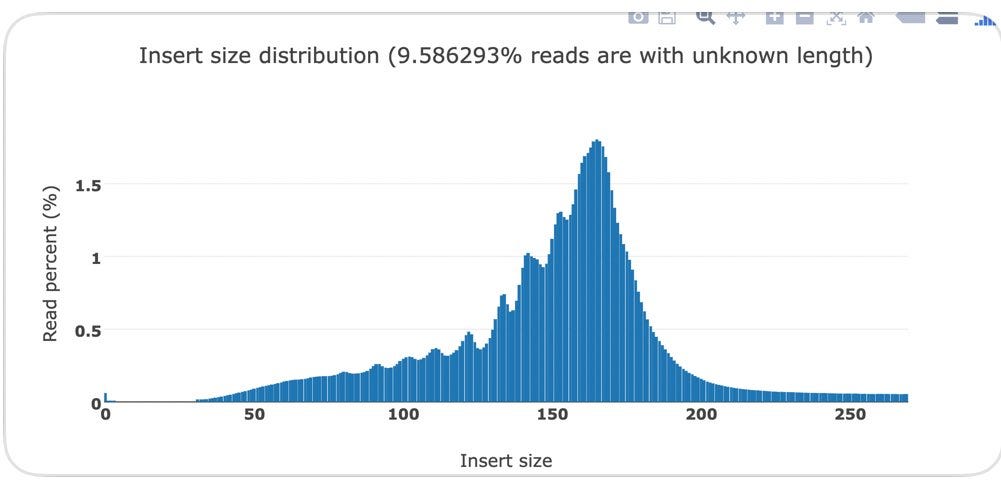
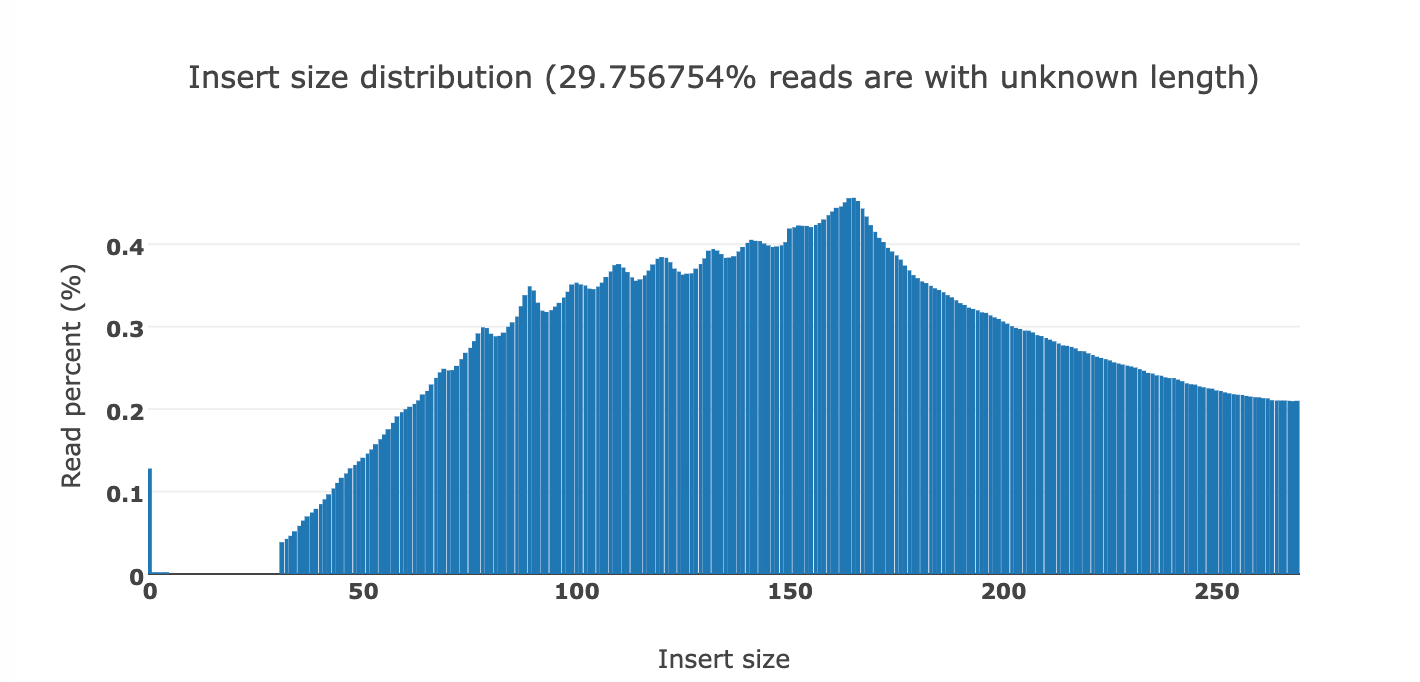
You will notice a ‘saw tooth’ in the insert size distributions that is modulus 10.5. Each of those peaks are 10-11 bases apart which is indicative of this DNA being nucleosome wrapped and under physical constraints of helical DNA being wrapped around a histone. The pitch of DNA is 10.5 bases per turn so this physical characteristic can be observed in DNA that needs to be tightly coiled around a histone. DNases that often attack cfDNA can only digest the linker DNA and fail to digest the histone wrapped DNA and the physical constraints of wrapping DNA gets reflected in the nuclease digestion patterns.

This fragmentation pattern is seen in other cfDNA studies exploring NETs.
These sequencing reads exhibit a fragmentation pattern consistent with NETs. There are 620M reads that map to human in the White clot (65X coverage) and 480M reads that map to human in the red clot (52X coverage). The unclassified reads are repetitive portions of the human genome that fail short k-mer (~32mers) classification with tools like KRAKEN.


If you take 10,000 150 bp reads and BLAST them as full length reads instead of short k-mers to Human, fewer reads will be unclassified.
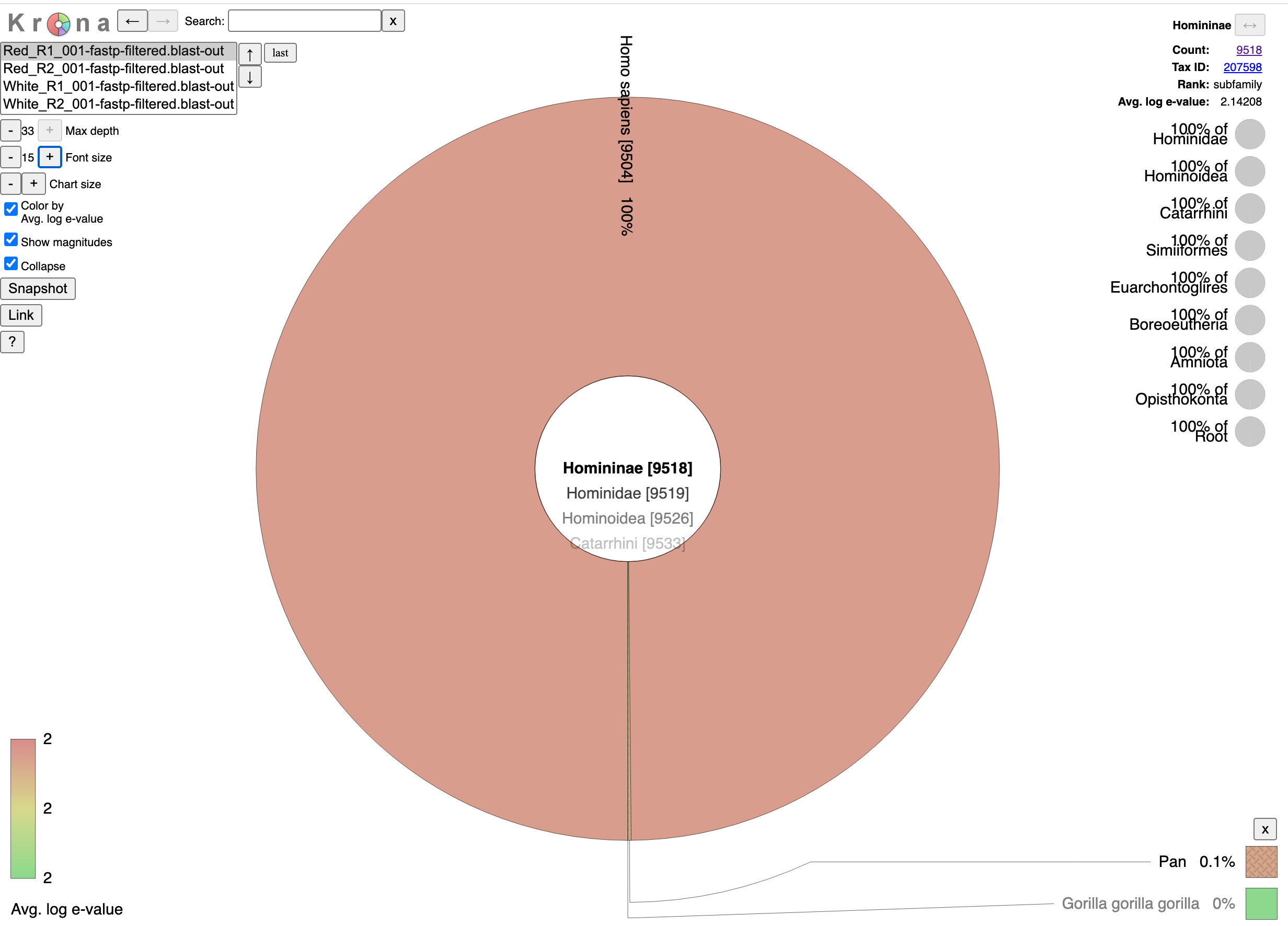
If you zoom in on the bacteria found in the sample you can see bacteria often found in vascular events.




The samples appear to be legitimate human derived clots with signs of NETs.
The next question to explore is if these patients have any DNA variants in the clotting cascade that may point to a genetic predisposition to clotting? There are 3 categories of genes we focus on: 27 core clotting and thrombosis related genes, 2312 genes with gene ontology terms (GO terms) that link them to the clotting and thrombosis cascade, and 201 genes with GO terms related to amyloidogenesis.


There does NOT appear to be Pfizer or Moderna sequence in the Red or White clots. We do not have information on the time from vaccination to clot or death.
Reads that do map to Pfizer or Moderna are of poor quality with many soft-clipped reads and mismatches. Since there is no spike sequence detected, we have not searched these reads for integration events. The mostly likely regions for integration are 3’ and 5’ UTR which share human sequences and demonstrate the read pile ups you see below (Figure 8). Those require careful analysis and we don’t expect to see integration events at 50X coverage in a clot unless they were recently vaccinated. Since the vaccines contain human sequences in their UTRs, teasing apart integration events from library construction artifact can be challenging and requires further investigation.
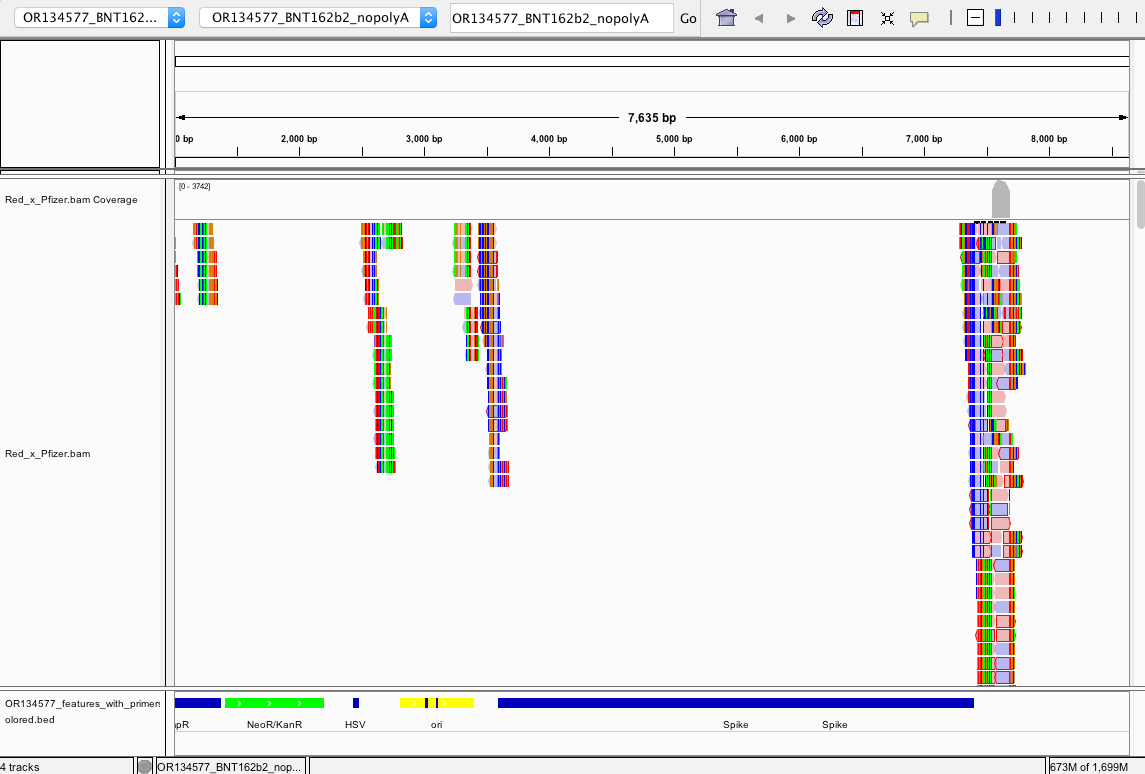
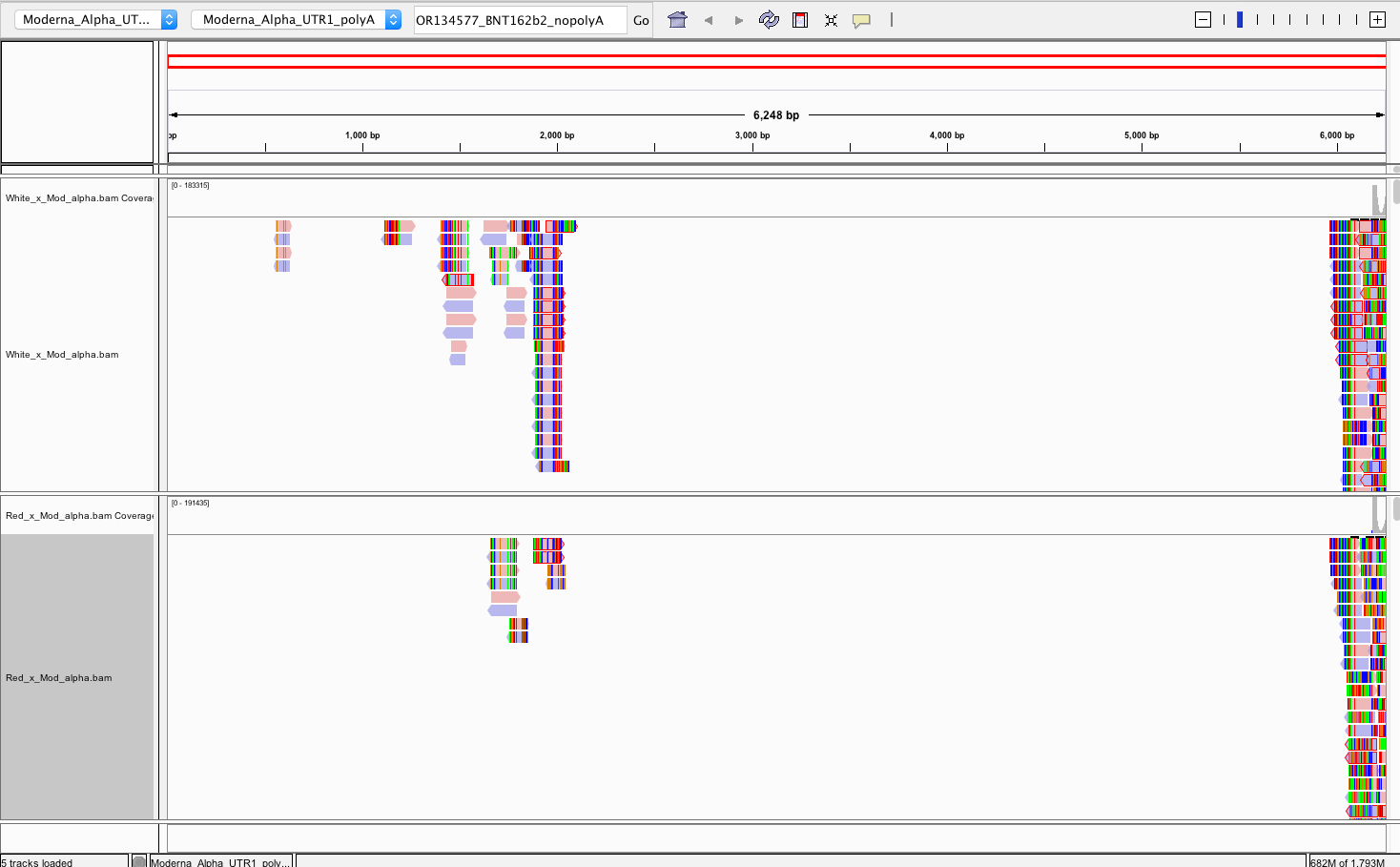
The snpEFF reports in the 27 core thrombosis related genes do display a list of MODERATE mutations of concern. In particular Factor V has 4 amino acid changing variants in the same gene some of which have been studied for inherited thrombophilia. Another missense mutation is also found in Factor XIII B subunit which is involved in Fibrin stabilization.
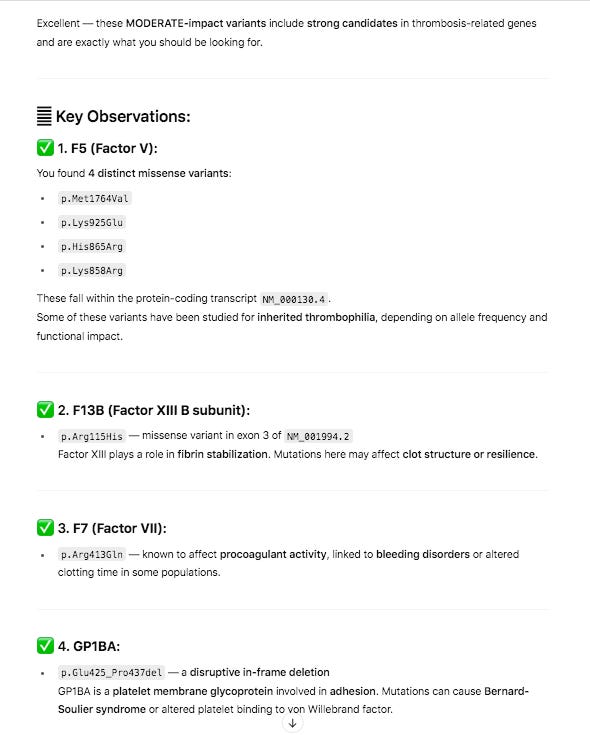
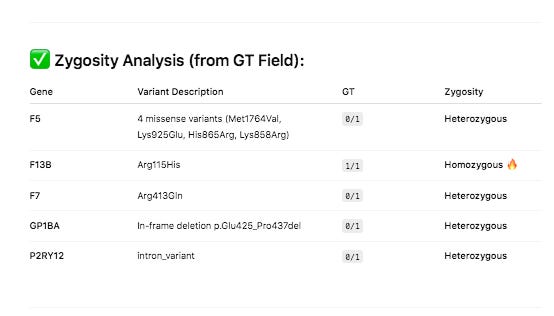
PolyPhen and SIFT measure each of these variants as ‘likely benign’. Neither tool assesses the impact of having 4 missense mutations in the same gene and if these can have a compounding effect. Short read Illumina data can phase two of the close variants (Lys858Arg and His865Arg) but the other two variants in F5 will need haplotype imputation to resolve.
Since the human genome is derived from such few ancestors, very few cross over events (recombination) have occurred across the chromosomes over time. As a result large blocks of the genome (haplotypes) are co-inherited throughout the population. Therefore, variants in close proximity to each other are informative on the likelihood of the identity of neighboring SNPs. This process is called phasing SNPs.

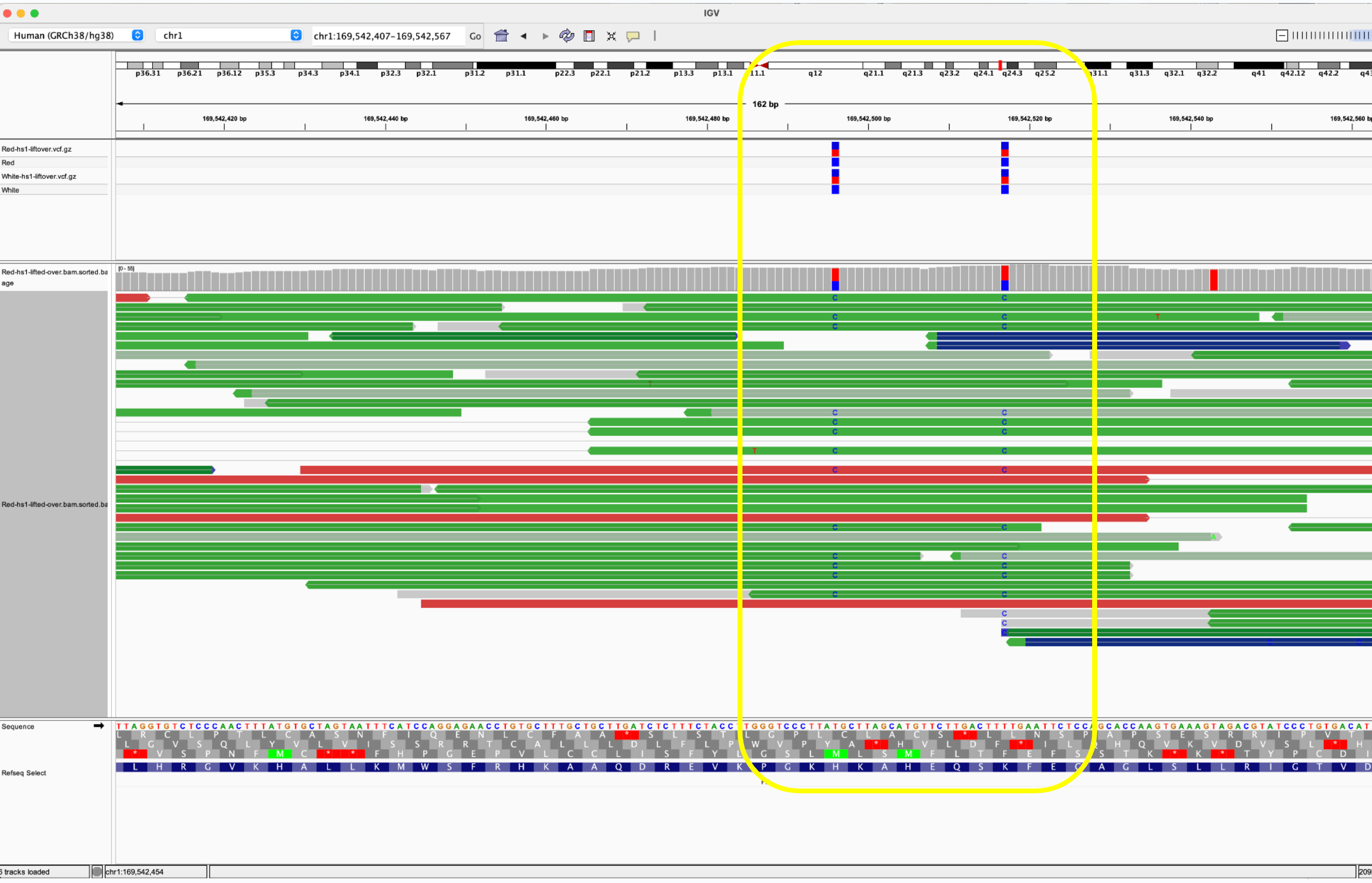
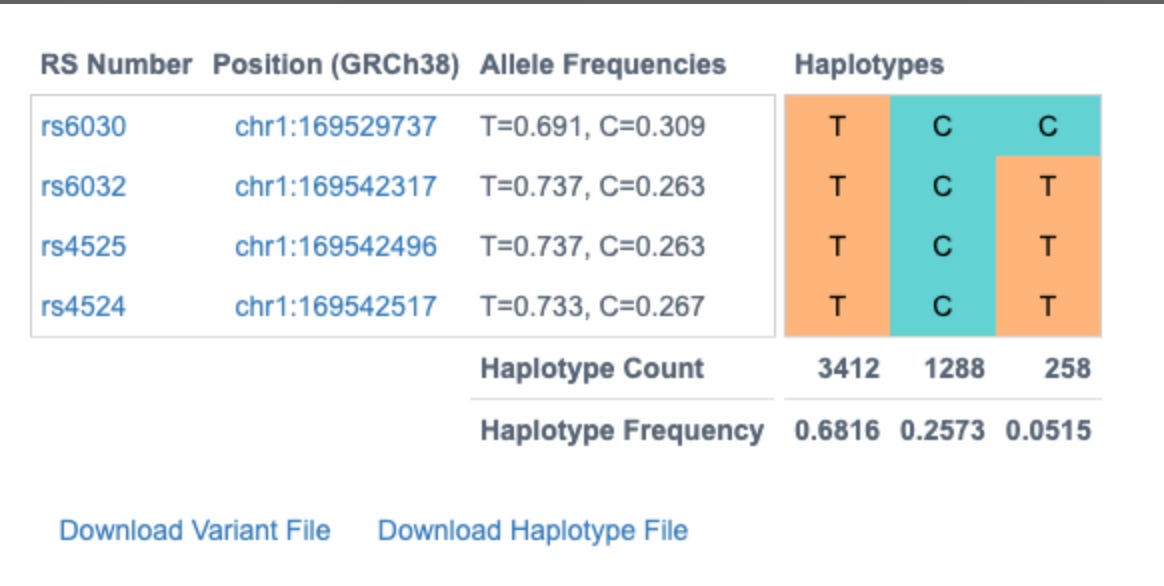
These 4 variants are known to exist in 3 dominant phases or haplogroups in the human population and we can infer which SNPs are riding on which chromosomes together. Knowing the phase of the variants is important for protein prediction as there are two copies of each gene.
Without knowing this phase we would have to calculate the structure of 2^4 or 16 different amino acids sequences. Phasing them leaves us with only 2 amino acid sequences to run predictions on and only these 2 biologically matter.
Expanding this analysis beyond the most published 27 genes in the clotting cascade, we can find 2312 gene that should be further scrutinized when using the following GO terms.

A list of all of the 2312 genes associated with these GO terms is here.
If you intersect the 2312 genes with the HIGH impact variants from snpEFF, we in fact, come up with more interesting hits that might be related to this phenomenon.
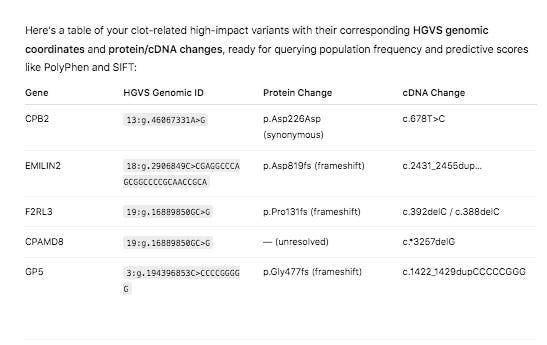

The variant in CPB2 is a silent mutation labelled by snpEFF as a ‘structural_interaction_variant’. There is evidence that this variant may play a role in RNA splicing.
The other 4 variants are Frameshift variants which are much more likely to be deleterious events.

Red Clot snpEFF HIGH impact variants in UniProt GO related genes.
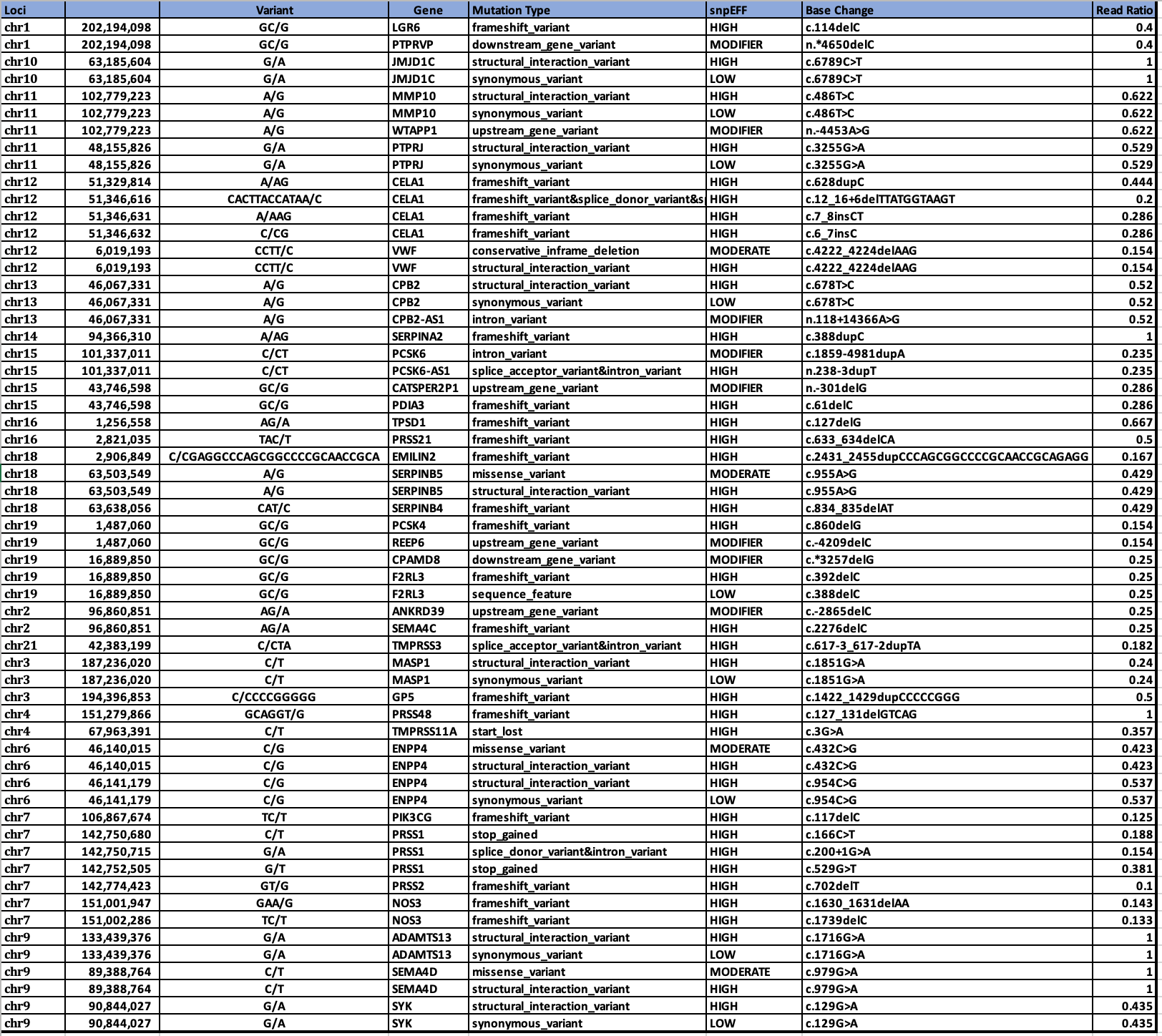
White Clot snpEFF HIGH impact variants in UniProt GO related genes.
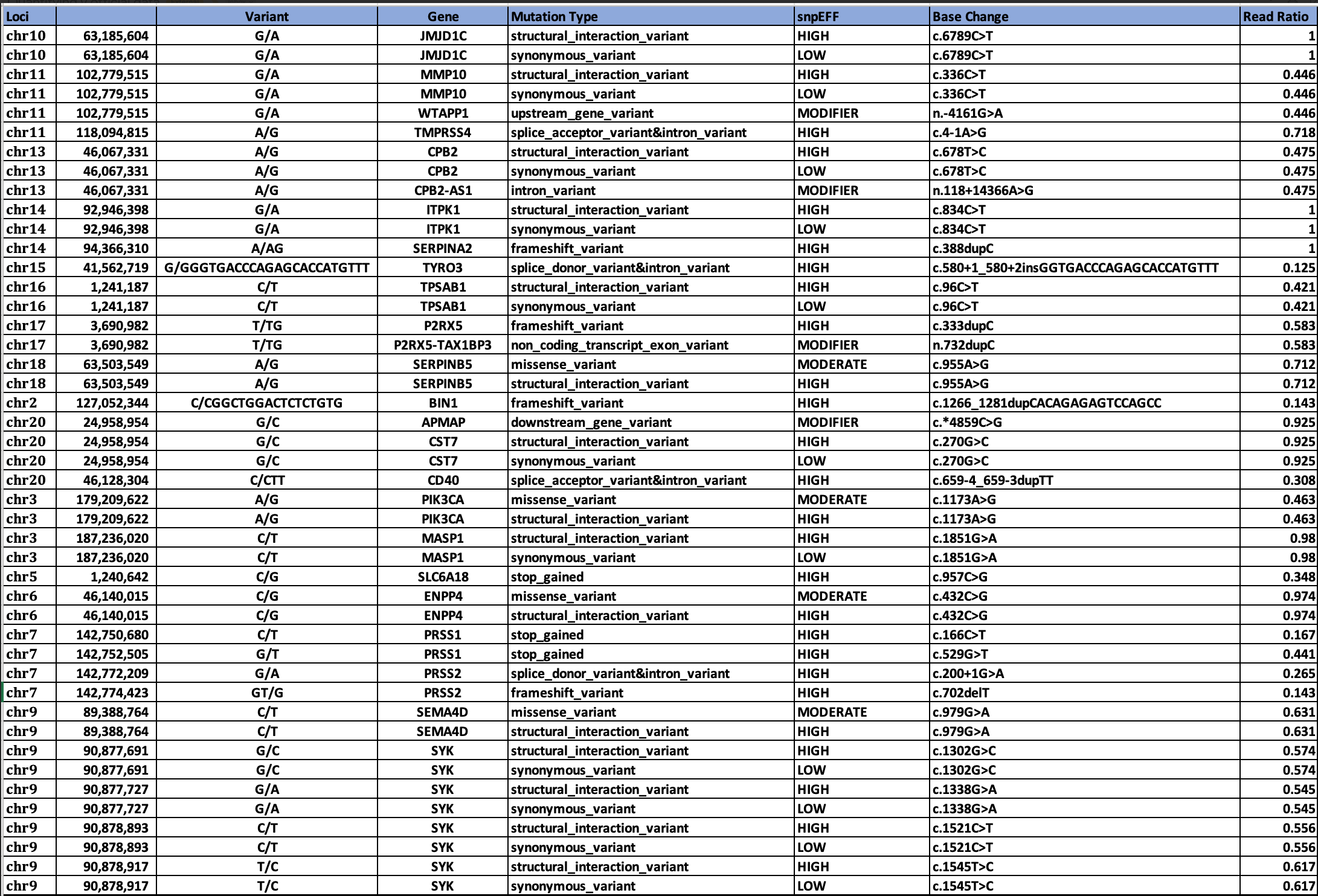
Genes in common between the two clots are SERPINs, CPB2, and MASP1.


We finally investigate 201 amyloidogenic genes with these GO terms for HIGH impact variants.
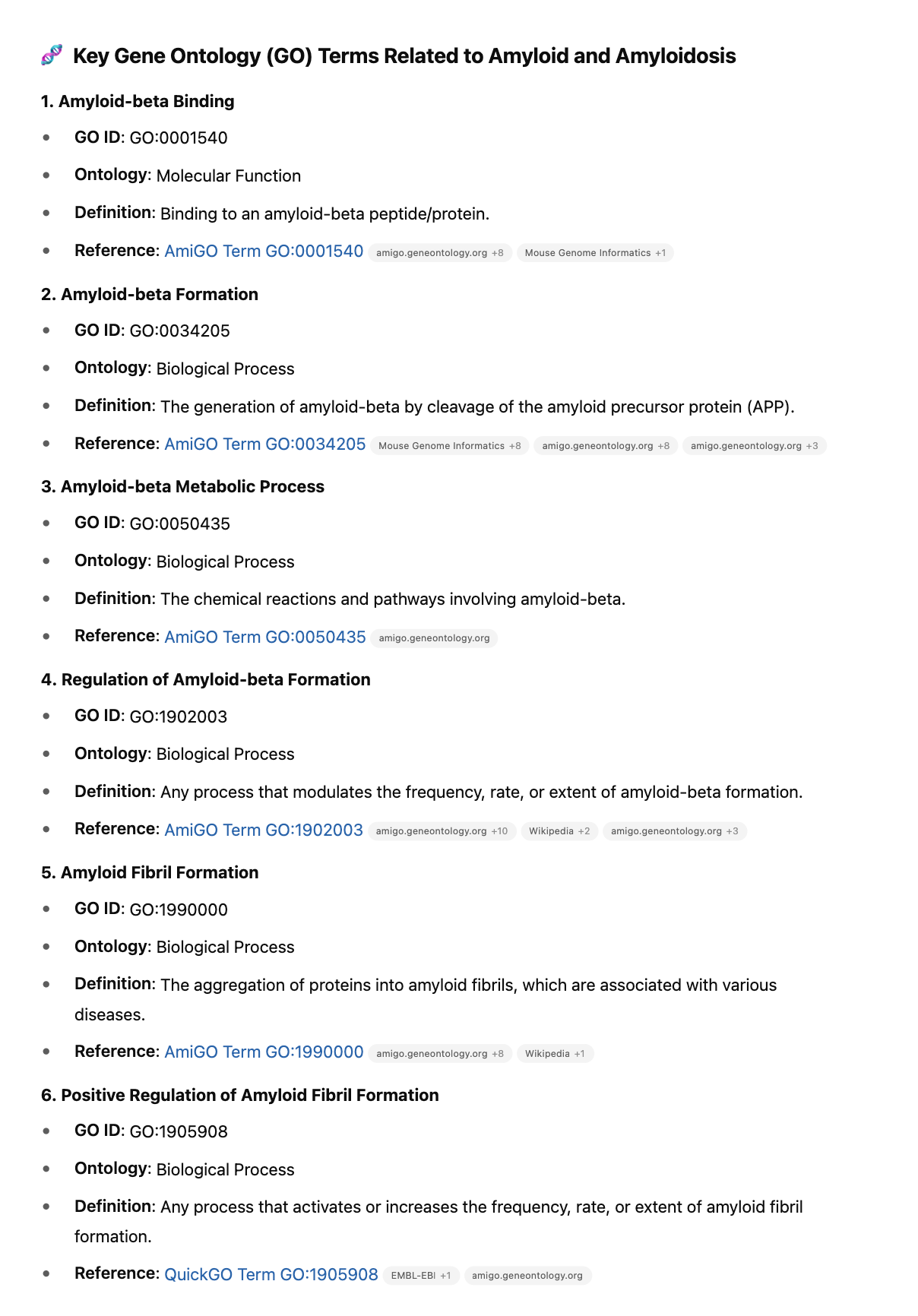
Red Clot variants in 201 amyloidogenic genes
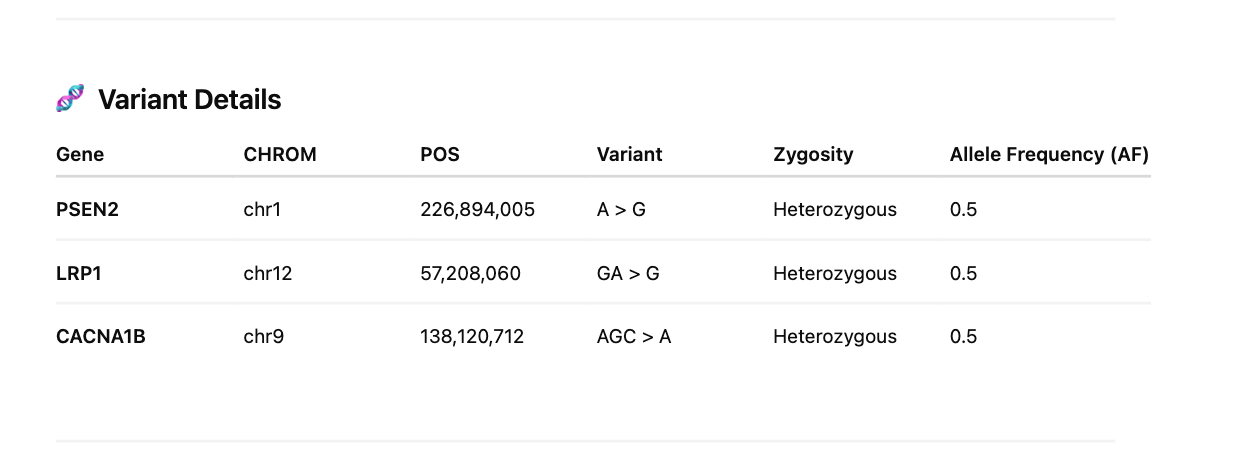

White Clot variants in 201 amyloidogenic genes

Discussion
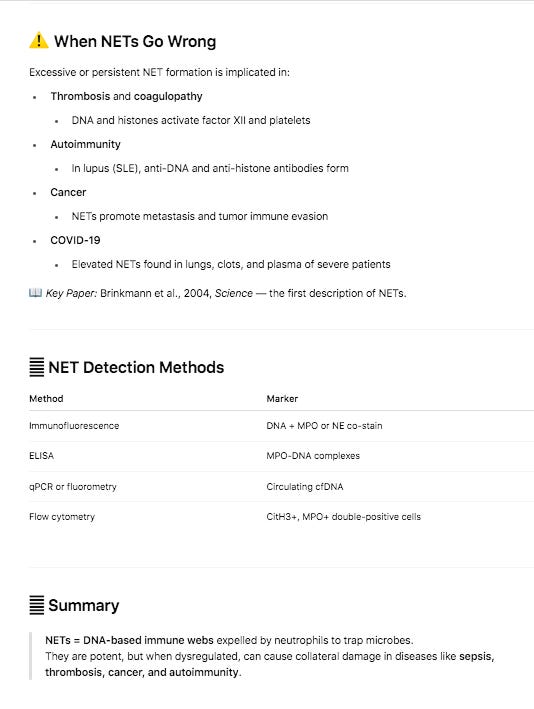
Sequencing of these clots exhibit signs of NETs often found in clotting reactions and confirmed they are in fact human samples. Highly damaging variants were found in genes involved in fibrinolysis (SERPINs, CPB2, MASP1) and amyloidogenesis (PSEN2 and LRP1). Other moderate variants were found in Factor V but these are far more speculative. More patients should be sequenced to assess if this association can be substantiated in larger cohorts and the population frequency of these damaging mutations further scrutinized.
In addition, during inquiry into the genetics of thrombotic predispositions, we must remain aware of the fact that the novel spike protein is amyloidogenic and prior variants associated with thrombotic predisposition may not capture this phenomenon. This study ‘looked under the lamp post’ by scanning genes involved in thrombosis and amyloidogenesis. The VCF files are available to enable the community to probe equally valid alternative hypothesis or gene sets.
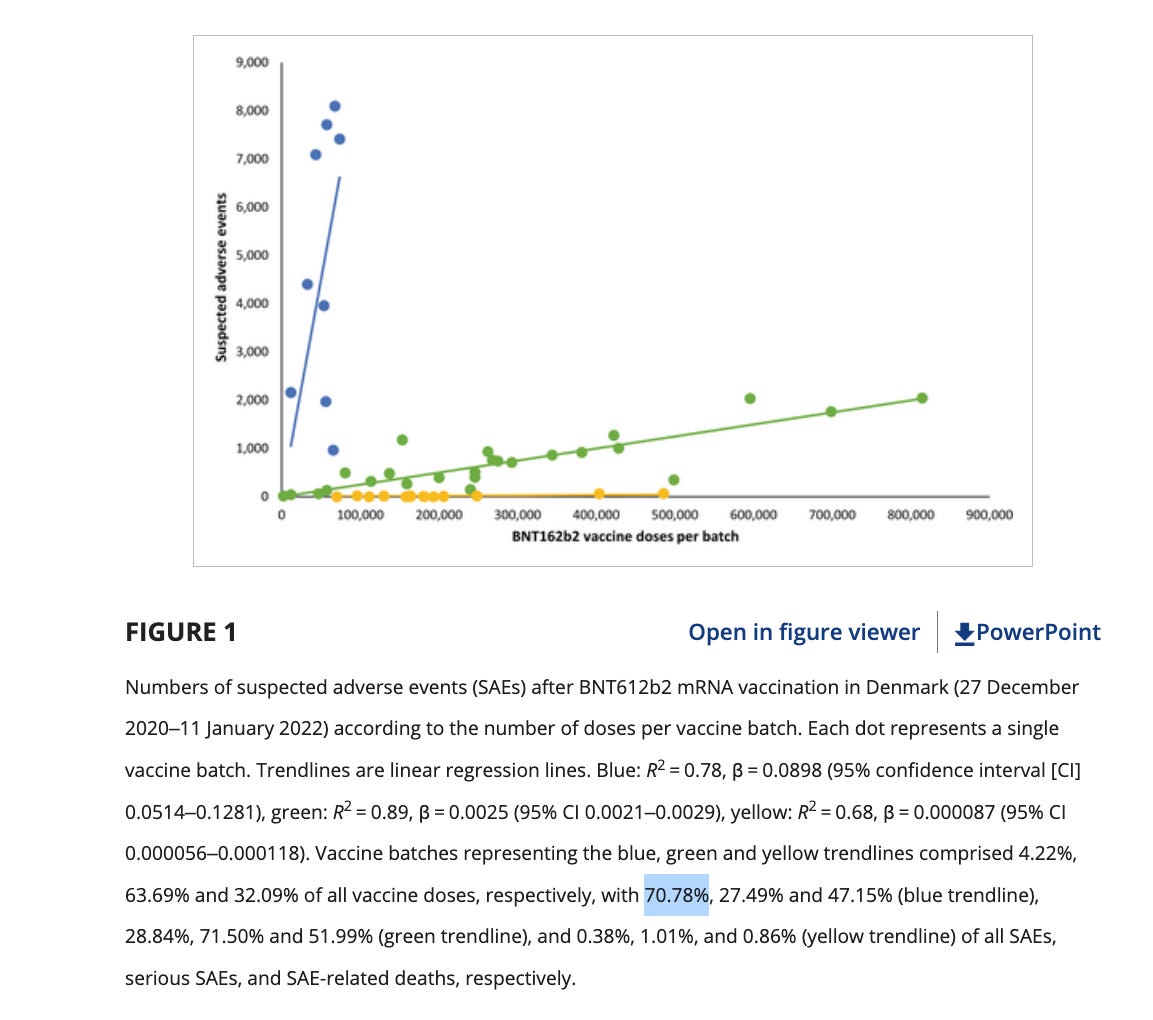
Further diluting this associative signal is the vaccine lot to lot heterogeneity as it pertains to severe adverse events. The Schmeling paper demonstrated that 4.22% of the lots accounted for 70% of the severe adverse events. Many people who have these genomic variants may escape a thrombotic event if administered a low adverse event lot. A combination of a bad lot with mutations in several genes related to amyloidogenesis, fibrinolysis or thrombotic pathways may be required to find a clean association of adverse events with these variants.
This is an N of 2 study and many more patients need to be sequenced to find such a novel association in a cohort where 95% of the lots showed decreased SAE.
Even with these limitations, this should be a ‘shot heard around the world’. Every patient who suffered a severe adverse event should be offered whole genome sequencing, RNA-Seq or both to understand what dysregulation is present and if this sequence data can inform more intelligent therapies.
This is a timely discussion as Vinay Prassad and Marty Makary are restricting the mRNA vaccine recommendations to only those patients at risk of C19. They should expand their scope to understand which patients are more at risk of mRNA adverse events. If they believe this is evidence based medicine, every risk/benefit analysis needs both a numerator and a denominator for BOTH the vaccine and the virus. The updated recommendations do not appear to have considered the currently unpredictable nature of who these vaccine adverse events strike. It is not just an age gradient as seen with the virus and the elderly people most at risk of C19 are likely equally or even more at risk of the vaccine. Their recommendations may turn ‘focused protection’ into ‘focused destruction’ if not properly informed.
Genetic predisposition to vaccine injury has not been thoroughly investigated to date and a genome sequencing program would illuminate if such predispositions exist, and if these variants can shed a light on the pathways of harm.
There are over 6 million C19 genomes sequenced in NCBI and many studies performed on the genomics of C19 susceptibility. Why is this the first one performed on the vaccine injured? Why is this being performed on a shoestring budget funded by substack subscriptions from autodidacts curious about this topic?
As a closing example: if you ask ChatGPT4.o what drugs one would consider for a SERPIN frameshift or Loss of Function mutation it has a few anticoagulants to suggest. This is obvious to most cardiologists. It also points to Colchicine to limit neutrophil activation and the described A1AT replacement therapy is probably not on anyones radar.

Conclusions
The clots removed from the vaccinated are in fact human, show evidence of Neutrophil Extracellular Traps and have HIGH impact genomic variants in the clotting and amyloidogenesis cascade. We cannot claim these variants are in fact causative of such adverse events. Its equally challenging to justify having vaccines with such an extraordinary adverse event frequency and with such polymorphic adverse event profiles remaining on the market. In either case, the genetic predispositions of the injured or deceased may inform on the pathways of harm and potential treatments with those currently suffering from these injuries. We should be saving these biopsies, sequencing their RNA and DNA so we can begin to see patterns in larger cohorts. In this cohort of vaccine injured people, they often feel abandoned by the current health care system as there is no ICD10 code for their novel condition. They are frequently being mis-diagnosed with anxiety, idiosyncratic disease, or even Munchausen’s. They deserve answers. The NIH has collected over 1 billion dollars in vaccine royalty they should commit to sorting it out.
Acknowledgements
Kevin McCairn PhD
Richard Hirschman
Steve McLaughlin, PhD
Yvonne Helbert, MS
Grep command used for Search
zgrep -E ‘F2|F5|F7|F8|F9|F10|F11|F12|F13A1|F13B|ITGA2B|ITGB3|GP1BA|GP1BB|GP9|P2RY12|TBXA2R|PROC|PROS1|SERPINC1|TFPI|PLAT|PLG|SERPINE1|FGA|FGB|FGG’ \
Red-hs1-liftover-to-hg38.vcf.snpEff.ann.MODIFIER-IMPACT.vcf.gz > clotting_genes.vcf
Illumina Sequencing Reads
BAM files mapped to Human Hs1 Reference and then lifted over to hg38 for better annotation.
Mapping Stats- Reads were mapped to Hs1 and BAM files lifted over to legacy human references (hg38) that are better annotated.
Red
1230576736 + 0 in total (QC-passed reads + QC-failed reads)
11354302 + 0 secondary
0 + 0 supplementary
193422579 + 0 duplicates
1229073887 + 0 mapped (99.88%:N/A)
1219222434 + 0 paired in sequencing
609611217 + 0 read1
609611217 + 0 read2
1117763356 + 0 properly paired (91.68%:N/A)
1217416260 + 0 with itself and mate mapped
303325 + 0 singletons (0.02%:N/A)
7188170 + 0 with mate mapped to a different chr
3728443 + 0 with mate mapped to a different chr (mapQ>=5)
White
1615731401 + 0 in total (QC-passed reads + QC-failed reads)
25278639 + 0 secondary
0 + 0 supplementary
508426706 + 0 duplicates
1613319407 + 0 mapped (99.85%:N/A)
1590452762 + 0 paired in sequencing
795226381 + 0 read1
795226381 + 0 read2
1566433026 + 0 properly paired (98.49%:N/A)
1587600712 + 0 with itself and mate mapped
440056 + 0 singletons (0.03%:N/A)
9166482 + 0 with mate mapped to a different chr
4341818 + 0 with mate mapped to a different chr (mapQ>=5)
SNP analysis